Clinical Report: Evaluating Palopegteriparatide for Pediatric ADH1
Overview
This case study presents a 16-year-old boy with Autosomal Dominant Hypocalcemia Type 1 (ADH1) who showed significant improvement in symptoms and biochemical parameters after treatment with palopegteriparatide. The findings suggest that this long-acting PTH analog may be a viable option when conventional therapies fail, particularly in managing calcium and phosphate levels.
Background
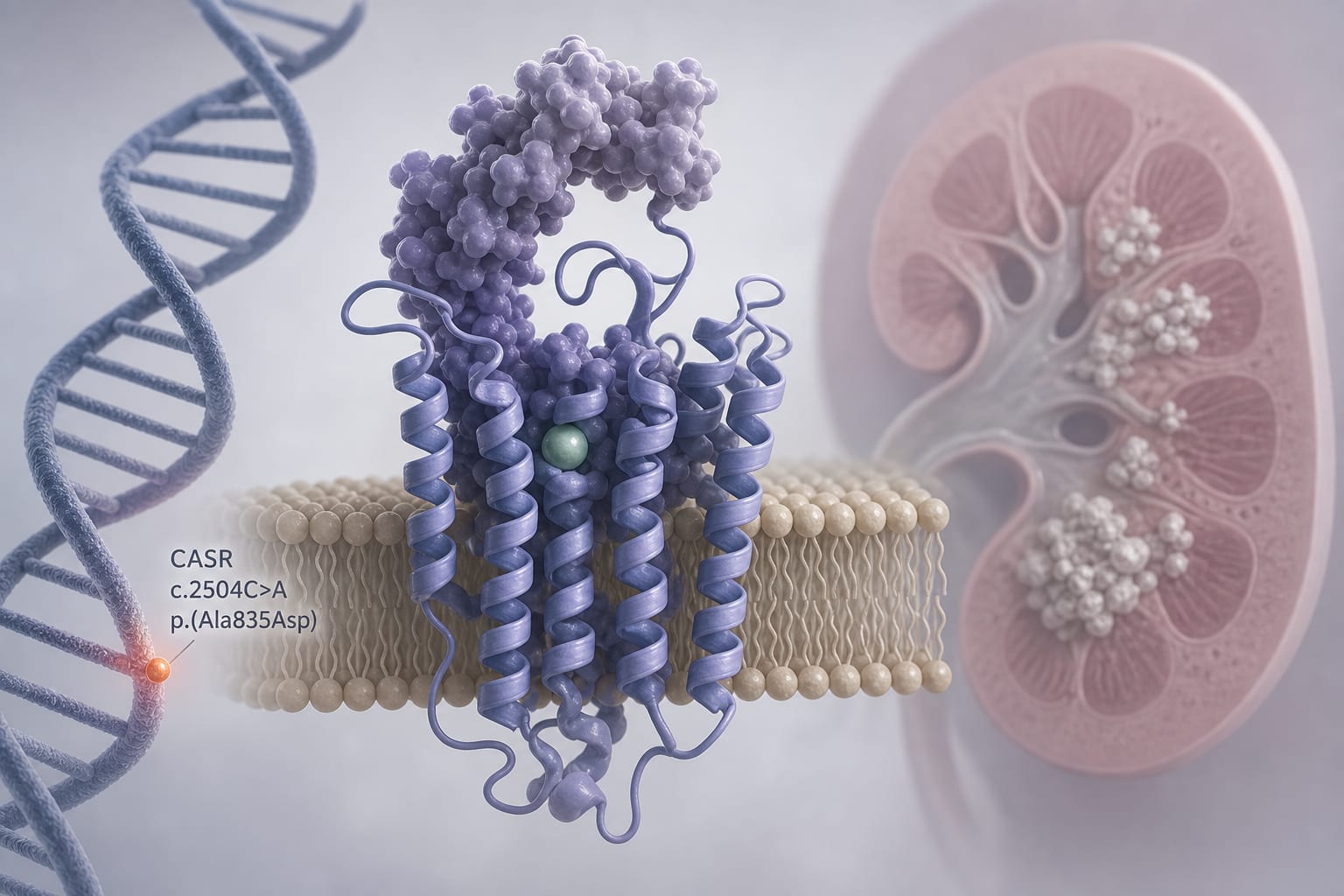
Autosomal dominant hypocalcemia type 1 (ADH1) is a rare genetic disorder linked to CASR gene mutations, resulting in severe calcium and phosphate imbalances. These mutations can lead to significant morbidity due to inadequate calcium regulation. Understanding new therapeutic options like palopegteriparatide is crucial for improving patient outcomes.
Data Highlights
No numerical data available in the article.
Key Findings
['A 16-year-old boy with a de novo CASR variant was diagnosed with ADH1.', 'Standard therapy with alfacalcidol and calcium supplements failed to control his symptoms.', 'Off-label treatment with palopegteriparatide led to complete resolution of tetany and improved biochemical stability.', 'Calcium and alfacalcidol supplementation were successfully discontinued after treatment.', 'This case highlights the potential of PTH analogs in managing pediatric ADH1.']
Clinical Implications
Pediatric patients with ADH1 may benefit from early consideration of PTH replacement therapy when conventional treatments are inadequate. Clinicians should monitor symptoms and biochemical parameters closely, including calcium and phosphate levels, to guide therapy adjustments.
Conclusion
Palopegteriparatide shows promise as a therapeutic option for pediatric ADH1, warranting further investigation into its long-term safety and efficacy, particularly in diverse patient populations.