Clinical Report: Uncommon KCNH2 p.D501N Mutation Linked to Early-Onset Malignant Long QT Syndrome
Overview
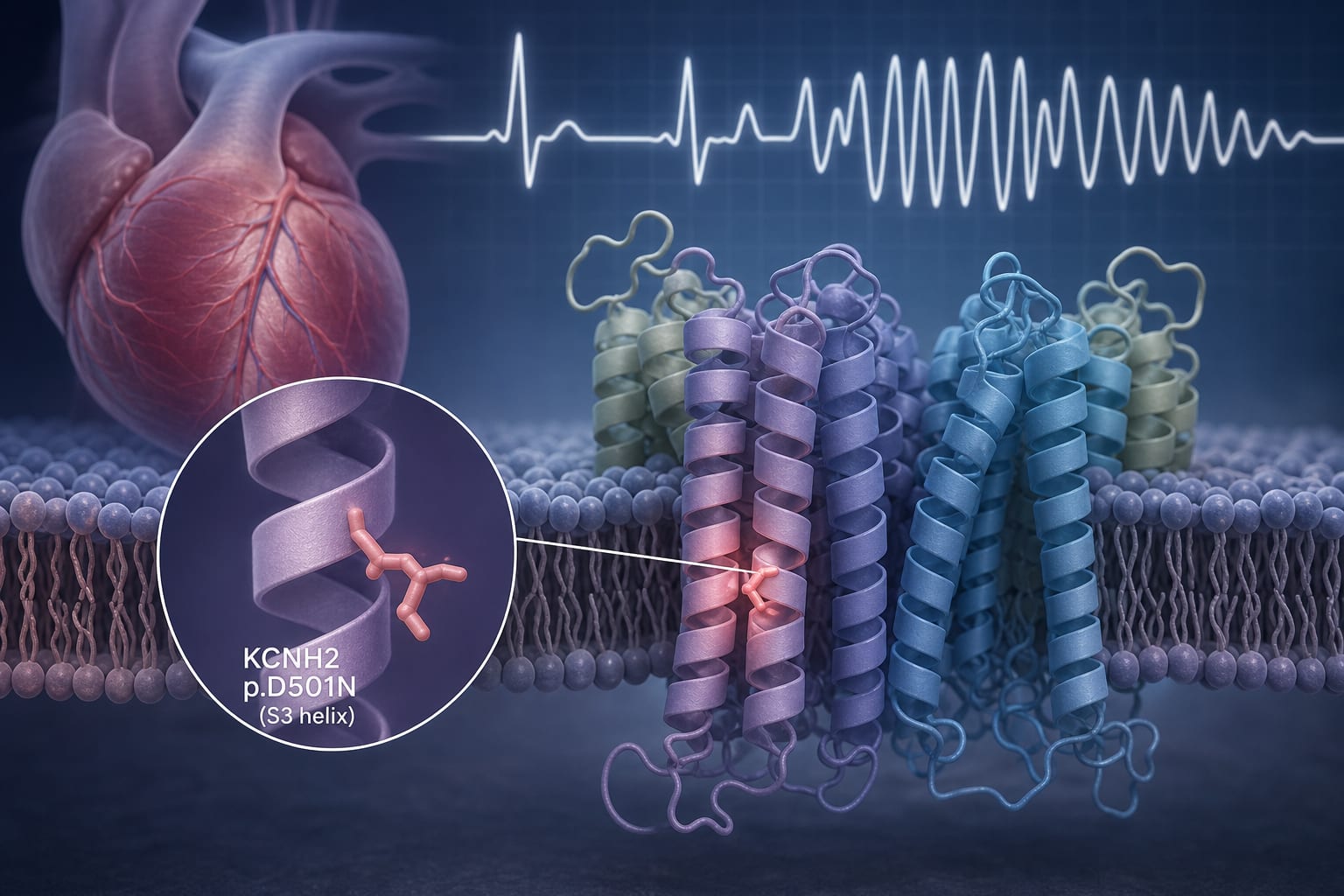
This report details a case of a 16-year-old girl with a de novo KCNH2 p.D501N mutation, presenting with severe long QT syndrome symptoms. The mutation significantly alters the Kv11.1 protein structure, leading to early-onset malignant LQT2.
Background
Long QT syndrome (LQTS) is a serious inherited channelopathy that can result in life-threatening arrhythmias and sudden cardiac death. It is primarily caused by mutations in ion channel genes, with KCNH2 mutations associated with LQT2. Understanding these mutations is crucial for early diagnosis and management of affected patients.
Data Highlights
No numerical data or trial data provided in the source material.
Key Findings
A 16-year-old girl with LQTS exhibited a prolonged QT interval and severe arrhythmias.
The patient carried a de novo KCNH2 p.D501N mutation, predicted to be deleterious.
This mutation alters the secondary structure of Kv11.1, increasing alpha helices and decreasing random coil.
KCNH2 p.D501N mutation leads to significant changes in the protein's physicochemical properties.
Management of the condition included ICD therapy, long-term β-blockers, and potassium–magnesium supplementation.
Clinical Implications
The identification of the KCNH2 p.D501N mutation emphasizes the importance of genetic testing in patients with unexplained LQTS. Early intervention with appropriate therapies can be critical in managing the risks associated with this condition.
Conclusion
The KCNH2 p.D501N mutation is linked to early-onset malignant LQT2, highlighting the need for genetic screening in similar cases. Understanding the mutation's impact on cardiac function is essential for effective patient management.