Clinical Report: First Report of an Intracranial Neuroendocrine Tumor in ROHHAD Syndrome
Overview
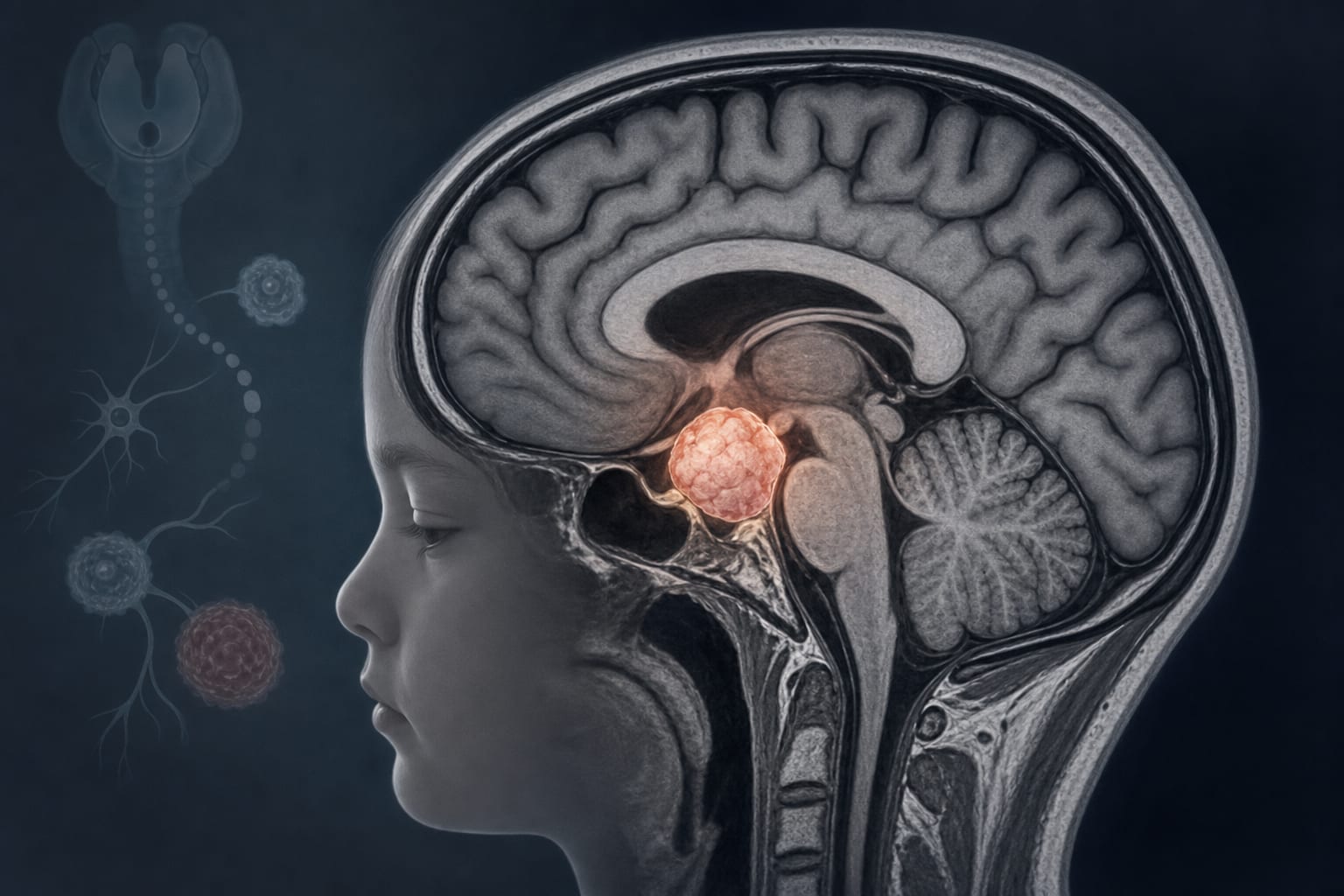
This report presents the first known case of an intracranial ganglion cell tumor in a patient with ROHHAD syndrome.
Background
ROHHAD syndrome is a rare pediatric disorder characterized by rapid-onset obesity, hypothalamic dysfunction, hypoventilation, and autonomic dysregulation. Approximately 40-50% of patients with ROHHAD develop associated neuroendocrine tumors, primarily ganglioneuroblastoma or ganglioneuroma.
Data Highlights
No numerical or trial data were provided in the source material.
Key Findings
This is the first report of an intracranial ganglion cell tumor in a patient with ROHHAD syndrome.
ROHHAD syndrome is associated with a significant risk of neuroendocrine tumors, with 40-50% of patients affected.
Diagnosis of ROHHAD syndrome is challenging due to the absence of a definitive diagnostic test.
Symptoms of ROHHAD syndrome can include rapid weight gain, hypoventilation, and autonomic dysregulation.
Clinical Implications
Healthcare providers should be vigilant in screening for both extracranial and intracranial tumors in patients presenting with symptoms of ROHHAD syndrome. Early identification and management of associated tumors may be crucial for improving patient outcomes.
Conclusion
This report presents a case of an intracranial tumor in a patient with ROHHAD syndrome.
by Nathalie J. Doelman-Oldenburger, Antoinette Y. N. Schouten-van Meeteren, Mariette E. G. Kranendonk, Kim Boshuisen, Michiel A. G. E. Bannier, Hanneke M. van Santen