Clinical Report: Mechanisms of Pathogenesis in Anderson-Fabry Disease
Overview
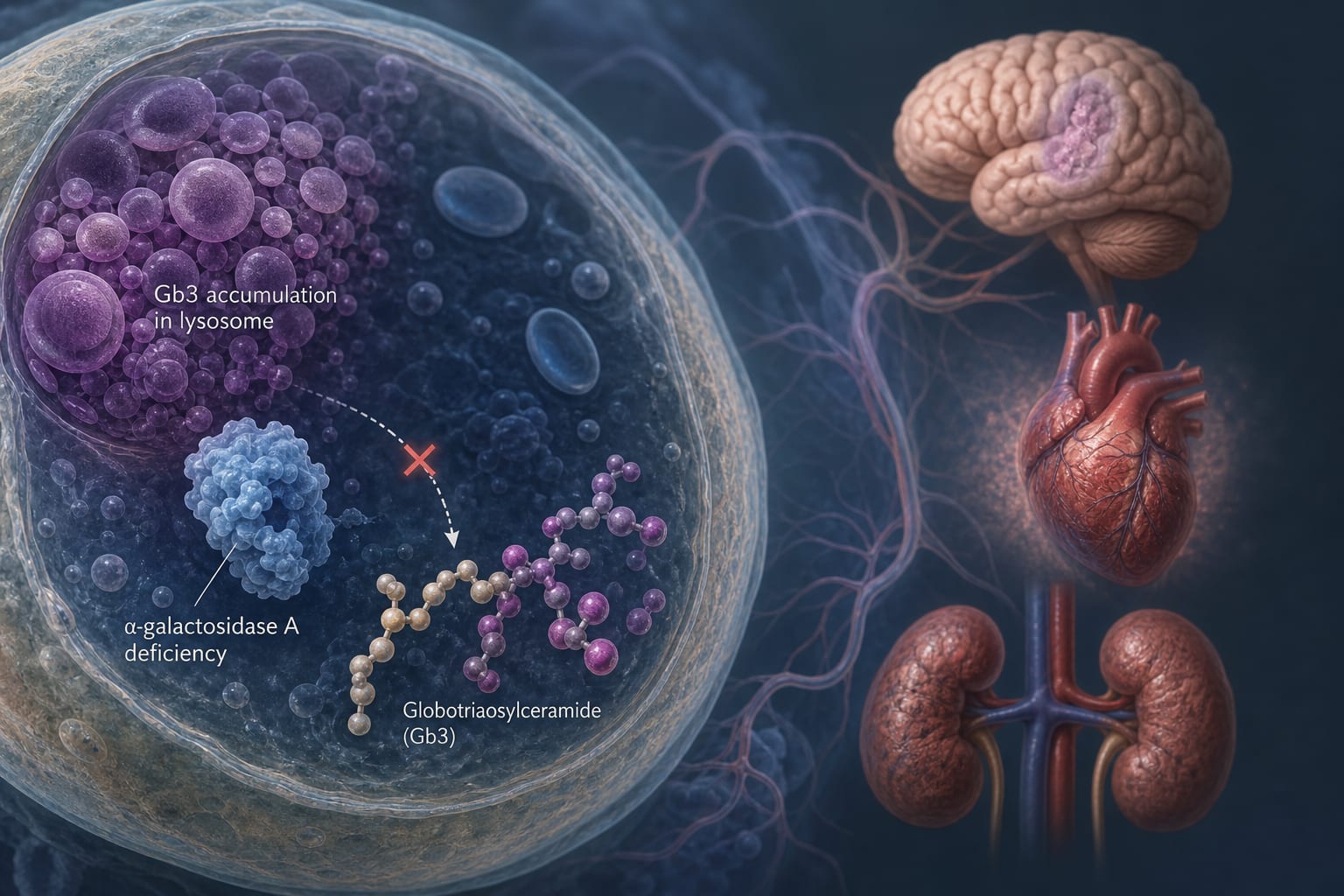
Anderson-Fabry disease (FD) is an X-linked lysosomal storage disorder caused by α-galactosidase A deficiency, leading to glycosphingolipid accumulation. The disease manifests with significant phenotypic variability and multisystem involvement, particularly affecting the kidneys and heart.
Background
Anderson-Fabry disease is a rare genetic disorder that results in the accumulation of globotriaosylceramide (Gb3) due to insufficient enzyme activity. This accumulation leads to progressive damage across multiple organ systems.
Data Highlights
No numerical data available in the source material.
Key Findings
FD is caused by mutations in the GLA gene leading to α-galactosidase A deficiency.
Phenotypic variability is influenced by residual enzyme activity and X-chromosome inactivation.
Classic FD typically presents with severe symptoms in males, while females may experience milder manifestations.
Common symptoms include renal insufficiency, cardiovascular issues, and neurological symptoms.
Accumulation of Gb3 and lyso-Gb3 contributes to cellular toxicity and organ dysfunction.
Clinical Implications
Healthcare professionals should be aware of the multisystem involvement in FD and the variability in presentation between genders.
Conclusion
Anderson-Fabry disease presents significant clinical challenges due to its complex pathogenesis and phenotypic variability.