Clinical Report: Genetic and Clinical Characterization of 30 Pediatric Cases with Gitelman Syndrome
Overview
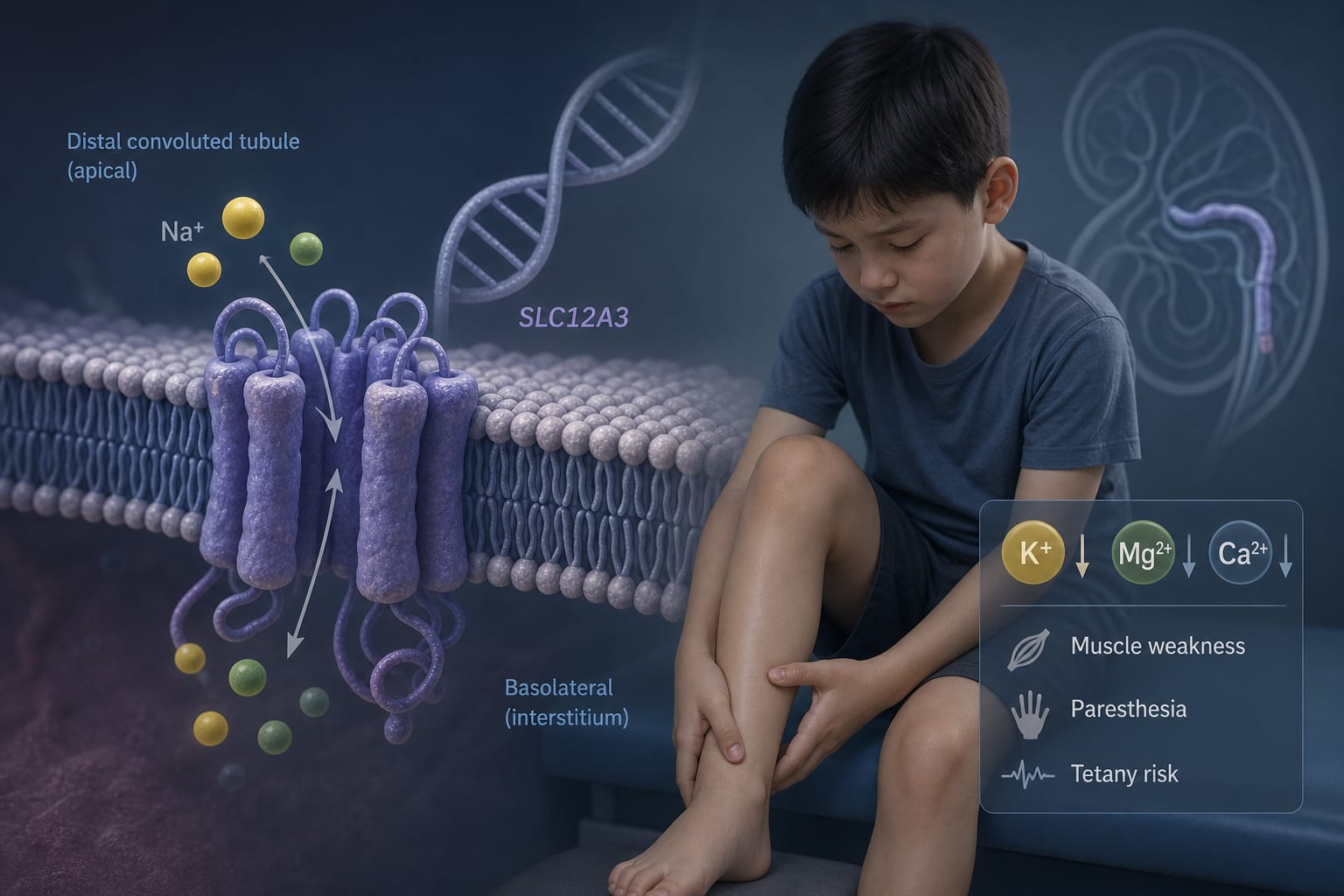
This study investigates the clinical phenotype and genetic spectrum of Gitelman syndrome (GS) in 30 pediatric cases in China. Key findings include a mean onset age of 7.9 years, common symptoms such as muscle weakness and hypomagnesemia, and the identification of 55 SLC12A3 variants.
Background
Gitelman syndrome is an autosomal recessive salt-losing tubulopathy caused by mutations in the SLC12A3 gene. It is characterized by significant clinical heterogeneity, which can lead to diagnostic challenges and long-term complications. Understanding the genotype-phenotype correlation is crucial for managing this condition effectively.
Data Highlights
Characteristic
Value
Mean onset age
7.9 ± 3.4 years
Muscle weakness
50%
Limb numbness
40%
Severe hypokalemia (<2.5 mmol/L)
Associated with tetany, dyslipidemia
Hypomagnesemia
90%
Identified SLC12A3 variants
55 (6 novel)
Key Findings
Mean onset age of Gitelman syndrome in the cohort was 7.9 years.
50% of patients presented with muscle weakness, and 40% with limb numbness.
All patients had hypokalemia, with 90% also experiencing hypomagnesemia.
Severe hypokalemia was linked to tetany and dyslipidemia.
55 SLC12A3 variants were identified, including 6 novel mutations.
No phenotypic differences were observed based on functional domain classification of mutations.
Clinical Implications
The findings highlight the importance of early screening for Gitelman syndrome in pediatric populations to manage symptoms effectively. Long-term follow-up is essential due to the potential for significant metabolic complications.
Conclusion
Pediatric Gitelman syndrome exhibits considerable clinical variability, and the diverse SLC12A3 mutations do not predict clinical outcomes based solely on functional domains. Ongoing research is needed to further elucidate genotype-phenotype relationships.