Pathogenic mechanisms in Fabry disease
By
Siming Wang
Chengyue Sun
June 23, 2026
Clinical Scorecard: Mechanisms of Pathogenesis in Anderson-Fabry Disease
At a Glance
Category Detail
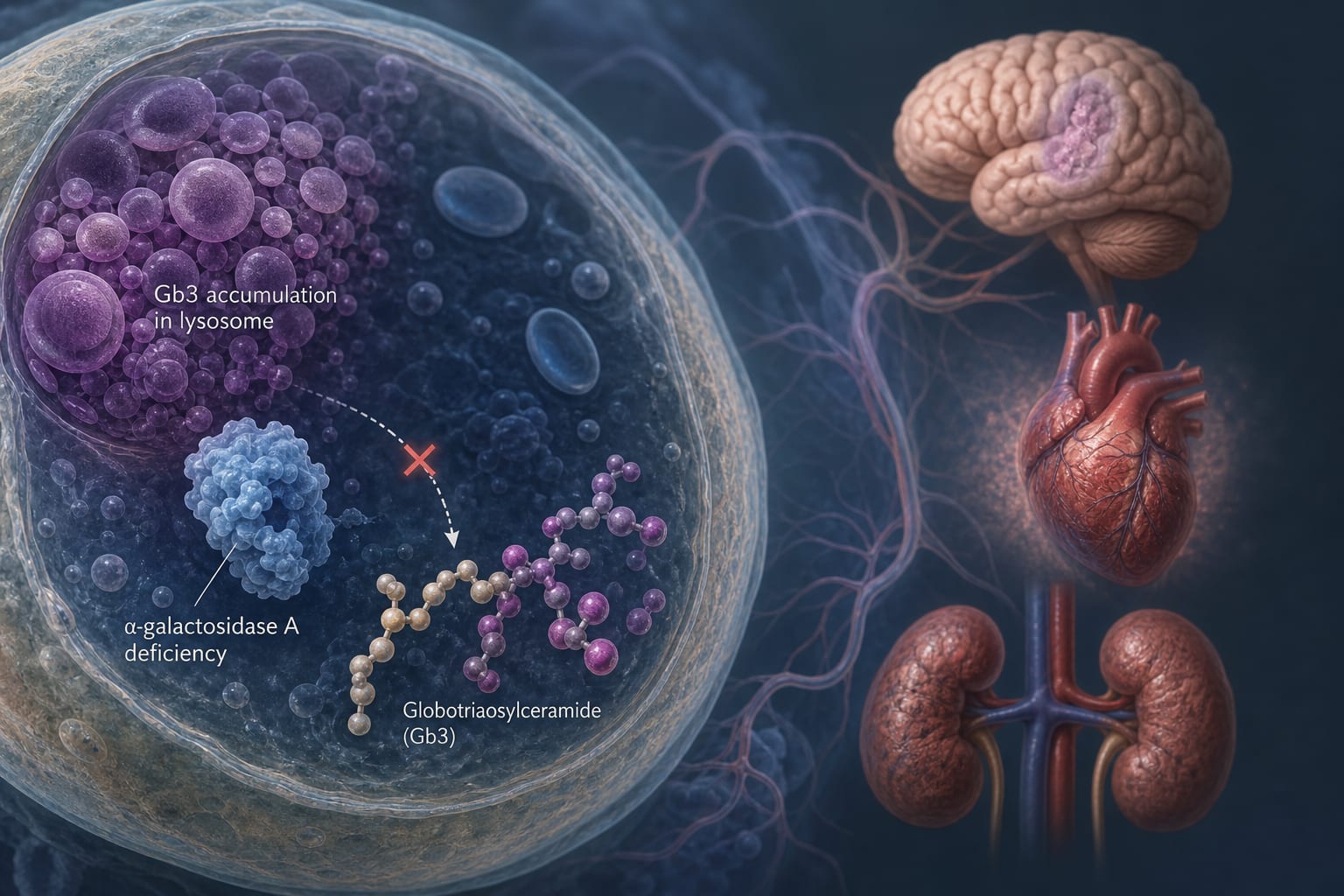
Condition Anderson-Fabry Disease Key Mechanisms Insufficient activity of α-galactosidase A leading to glycosphingolipid accumulation, lysosomal dysfunction, inflammation, and oxidative stress. Target Population Individuals with X-linked lysosomal storage disorder, particularly males and heterozygous females. Care Setting Clinical genetics and metabolic disorder management.
Key Highlights
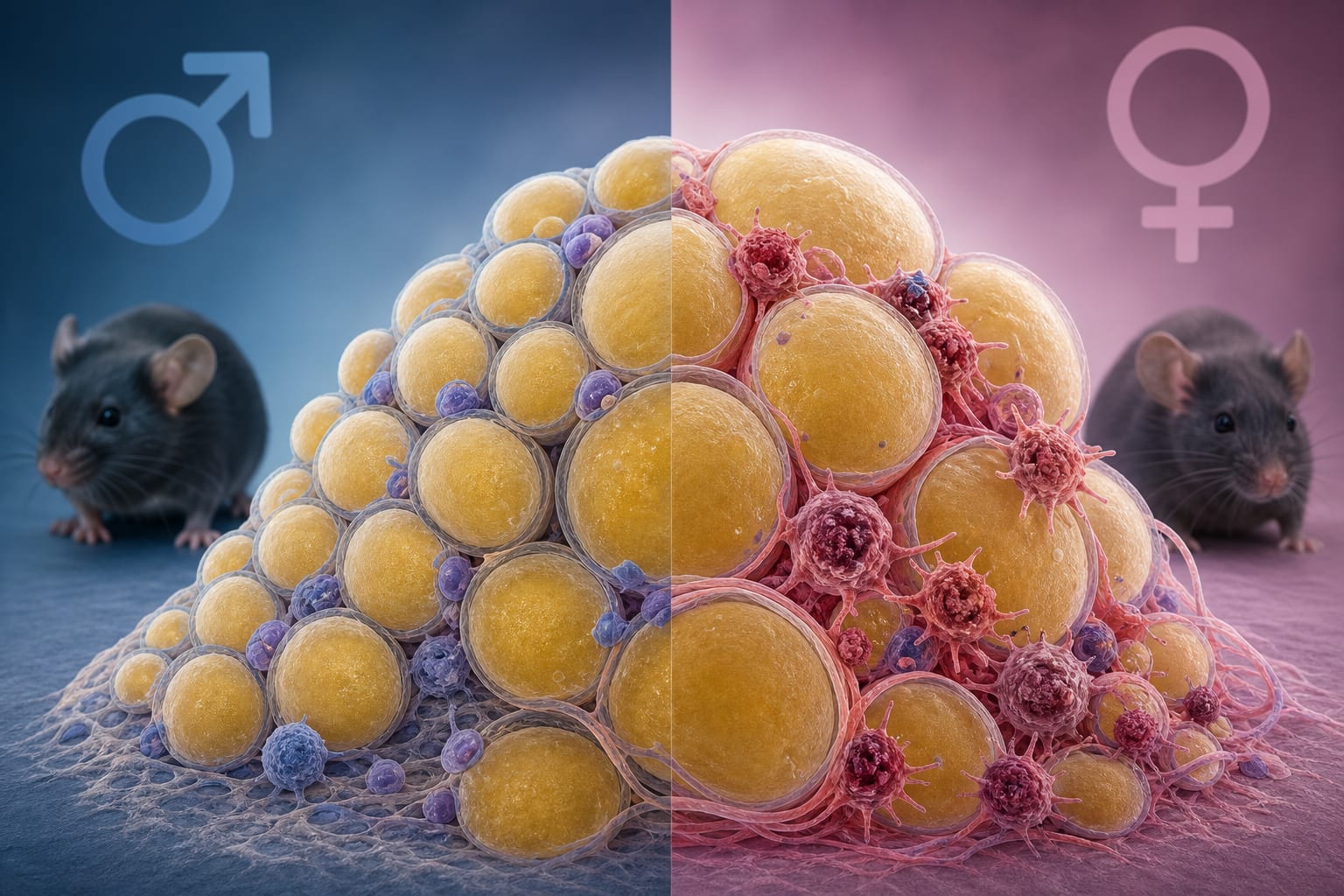
X-linked disorder caused by α-galactosidase A deficiency. Phenotypic variability influenced by residual enzyme activity and X-chromosome inactivation. Multisystem involvement includes cardiovascular, renal, and neurological manifestations. Life expectancy significantly reduced, particularly in males. ER stress and UPR activation contribute to disease variability. Guideline-Based Recommendations
Diagnosis
Diagnosis based on enzyme activity measurement and genetic testing. Management
Enzyme replacement therapy and symptomatic management. Monitoring & Follow-up
Regular assessment of organ function and symptom progression. Risks
Increased risk of renal failure, cardiovascular events, and strokes. Patient & Prescribing Data
Males typically present with classic FD; females may exhibit variable symptoms.
Enzyme replacement therapy is the primary treatment option.
Clinical Best Practices
Early diagnosis and intervention to manage symptoms. Genetic counseling for affected families. Monitoring for cardiovascular and renal complications. Related Resources & Content