Clinical Scorecard: Case Study: Fabry Disease Presenting as Coronary Artery Disease and Hypertrophic Cardiomyopathy—A 15-Year Delay in Diagnosis
At a Glance
Category
Detail
Condition
Fabry Disease
Key Mechanisms
X-linked hereditary lysosomal storage disorder caused by mutations in the GLA gene leading to α-galactosidase A deficiency. Prevalence is approximately 1 in 100,000.
Target Population
Patients with unexplained left ventricular hypertrophy or coronary artery disease, particularly those with family history.
Care Setting
Cardiology and genetic testing facilities.
Key Highlights
Fabry disease can present as coronary artery disease or hypertrophic cardiomyopathy, complicating diagnosis.
Average diagnostic delay exceeds 10 years, often missing critical early intervention opportunities.
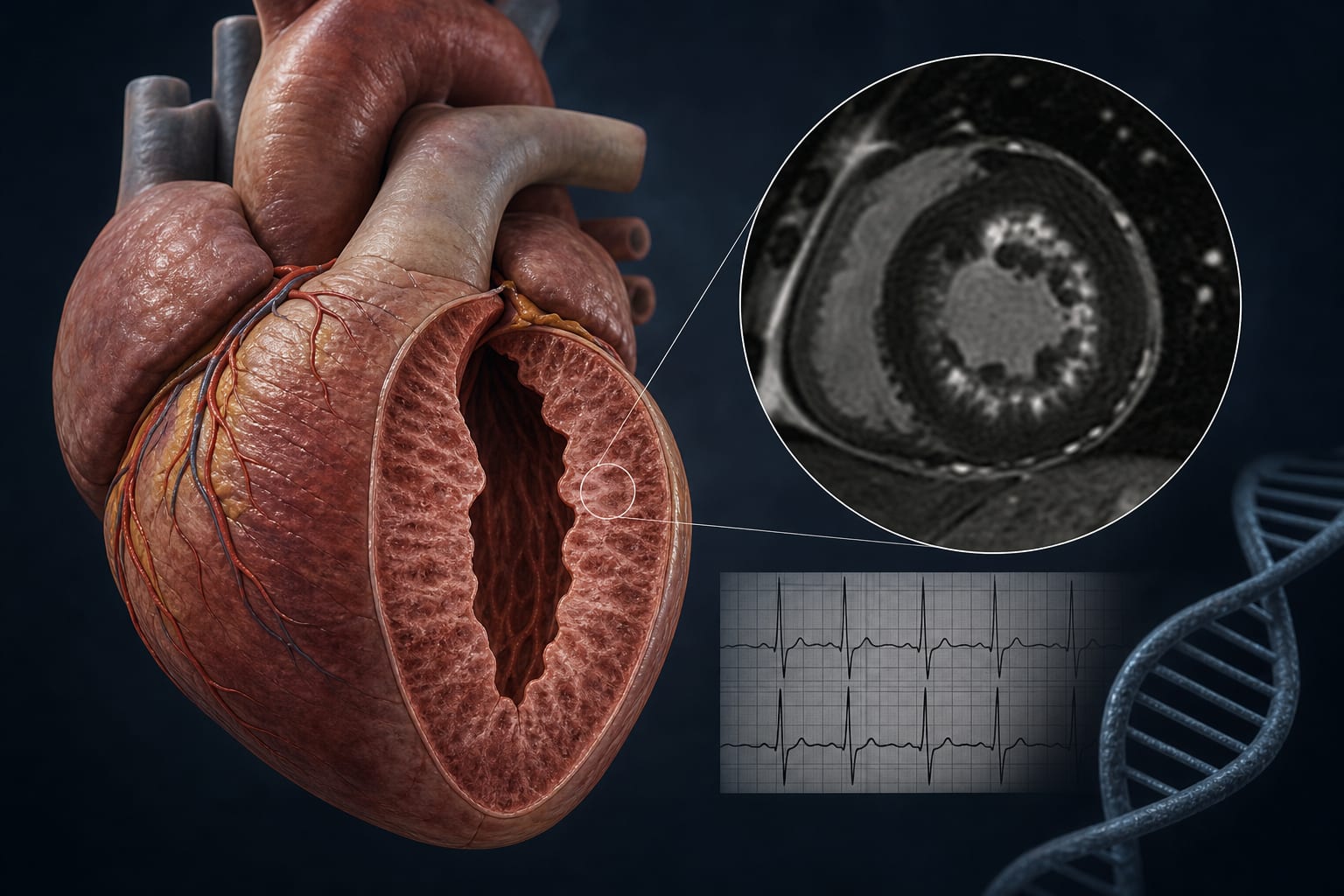
Characteristic imaging findings and enzymatic assays are crucial for diagnosis.
Genetic testing is essential for confirming Fabry disease.
Guideline-Based Recommendations
Diagnosis
Utilize clinical warning signs, cardiac magnetic resonance imaging, enzymatic assays, and genetic testing.
Management
Enzyme replacement therapy is recommended but may not always be initiated; consider patient-specific factors.
Monitoring & Follow-up
Regular assessment of cardiac function and symptoms in patients with suspected Fabry disease.
Risks
Increased risk of heart failure, malignant arrhythmias, stroke, and death due to delayed diagnosis.
Patient & Prescribing Data
Adult males with unexplained left ventricular hypertrophy or coronary artery disease.
Management includes heart failure and secondary prevention regimens for coronary artery disease, with consideration for enzyme replacement therapy.
Clinical Best Practices
Consider Fabry disease in differential diagnosis for adult-onset left ventricular hypertrophy.
Implement a diagnostic pathway to reduce misdiagnosis and missed diagnosis.
Encourage interdisciplinary collaboration for comprehensive patient evaluation.