To investigate the effects of Lipoprotein(a) [Lp(a)] on cardiomyocytes and its role in inducing ferroptosis.
Approach:
In vitro and in vivo studies: Utilized cell culture and small animal models to assess the impact of Lp(a) on cardiomyocytes.
Key Findings:
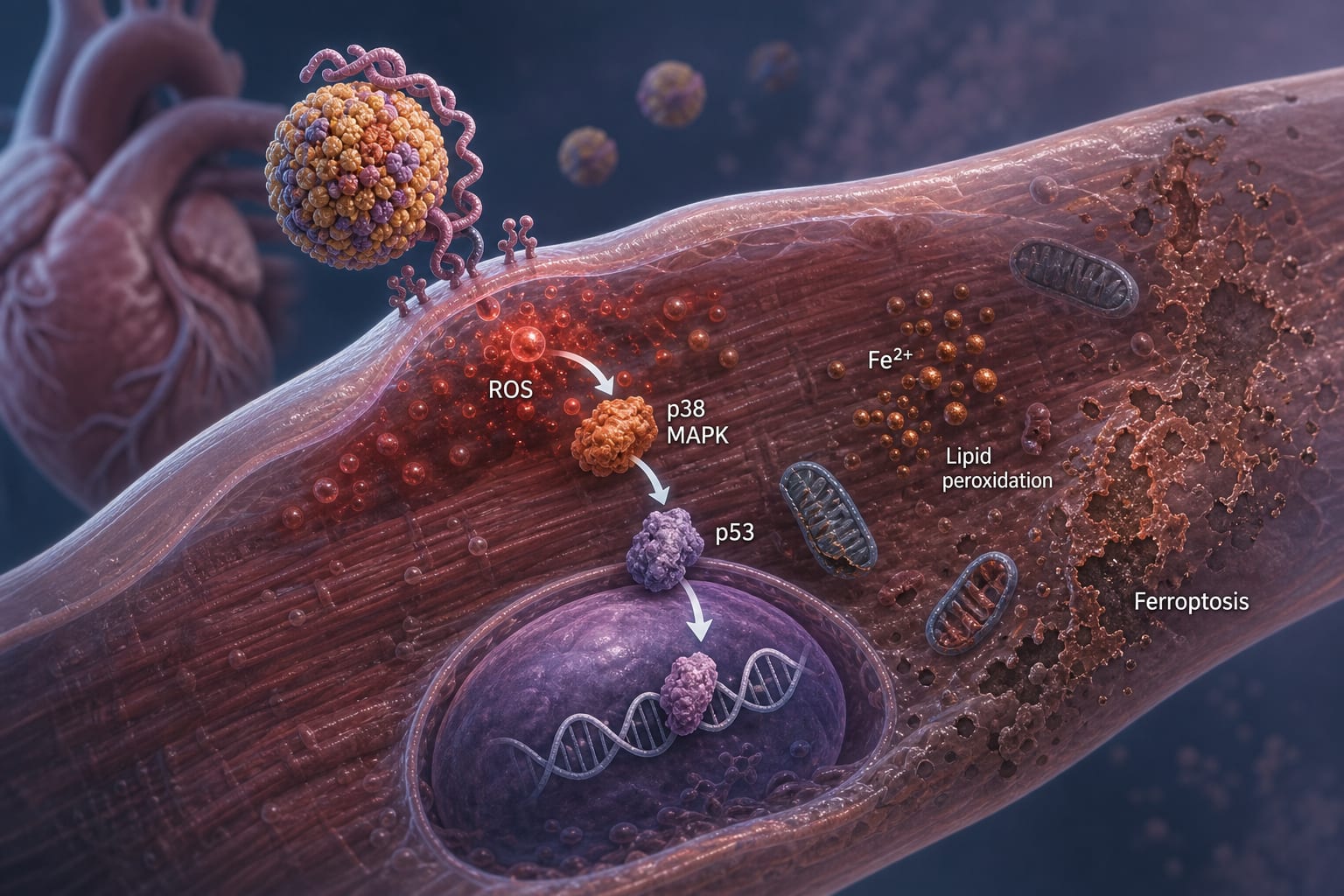
Lp(a) induces ferroptosis in cardiomyocytes via a redox-sensitive pathway involving p38 MAPK activation and p53-mediated transcriptional regulation.
Exposure to Lp(a) led to increased intracellular Fe2+ levels, elevated malondialdehyde (MDA), and phosphorylation of p38 MAPK.
Pharmacological blockade of p38 using SB203580 or siRNA-mediated p38 silencing significantly reduced ferroptotic markers.
Activation of p38 promotes nuclear translocation of p53, which suppresses SLC7A11 expression, contributing to ferroptosis.
In vivo studies in Lp(a)-treated C57BL/6J mice confirmed cardiac dysfunction and ferroptotic markers, including myocardial Fe2+/MDA elevation and glutathione/cysteine depletion.
Interpretation:
The study investigates the mechanisms by which Lp(a) contributes to cardiomyocyte injury through ferroptosis, focusing on the roles of p38 MAPK and p53.
Limitations:
The study primarily focuses on specific pathways and may not encompass all mechanisms of Lp(a) induced cardiomyocyte injury.
Further research is needed to explore the implications of these findings in clinical settings.
Conclusion:
Lp(a) activates p38 MAPK through increased ROS levels, promoting ferroptosis in cardiomyocytes via SLC7A11 inhibition dependent on p53 activation.