To consolidate insights into the molecular and cellular pathogenesis of Anderson-Fabry disease (FD) and mechanisms contributing to phenotypic variability.
Approach:
Key Findings:
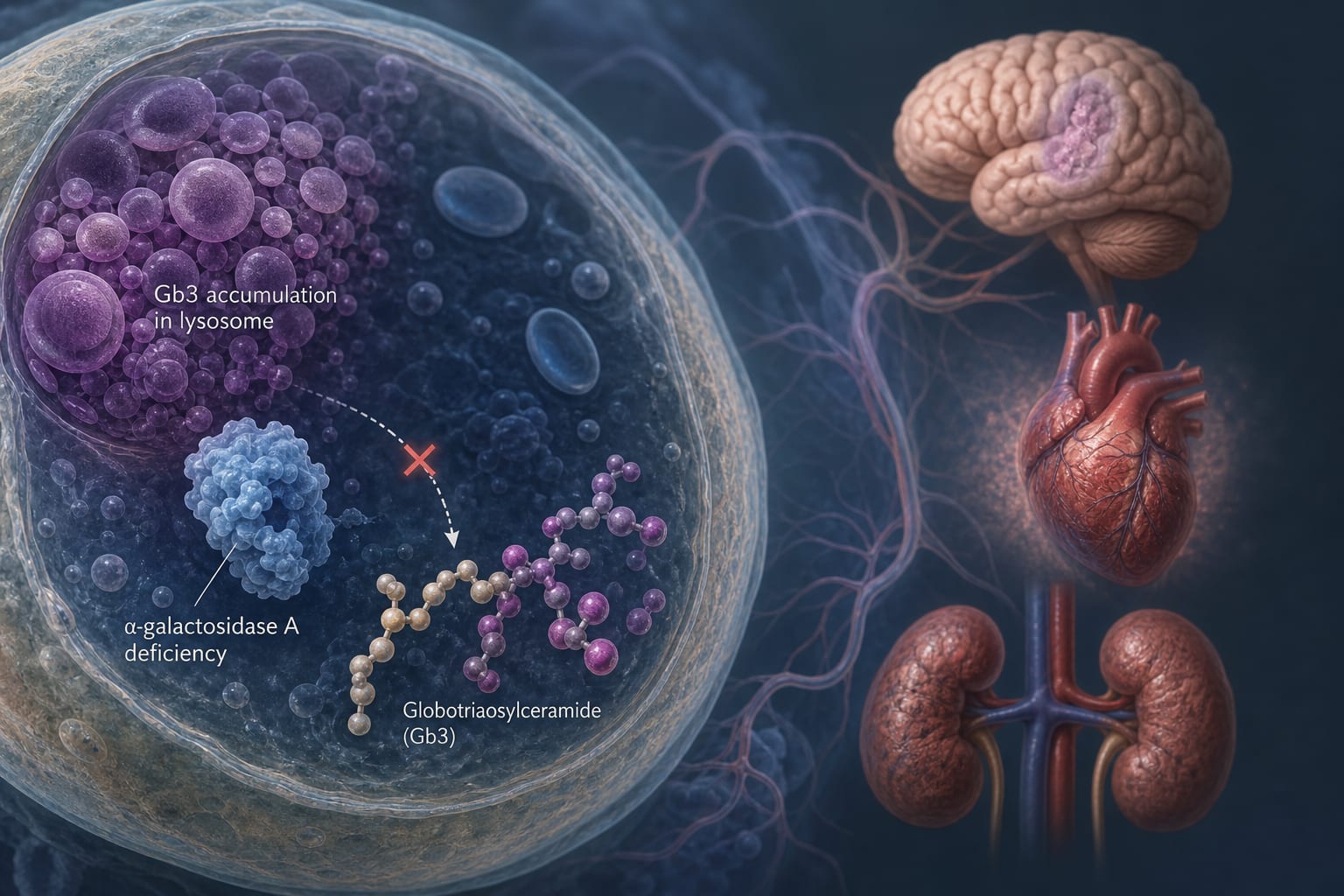
FD is caused by insufficient activity of α-galactosidase A, leading to the accumulation of glycosphingolipids, particularly Gb3.
Phenotypic variability is influenced by residual enzyme activity, type of mutation, and X-chromosome inactivation.
Classic FD typically presents earlier and more severely in males, while females exhibit a range of symptoms due to X-inactivation patterns.
Accumulation of Gb3 and lyso-Gb3 contributes to cellular toxicity and organ dysfunction.
Interpretation:
The review highlights the complexity of FD pathogenesis and factors contributing to clinical variability.
Limitations:
The review may not cover all recent studies or emerging insights into FD pathogenesis.
Current understanding of X-chromosome inactivation and enzyme activity may not fully explain phenotypic variability.
Conclusion:
The study provides an overview of the mechanisms underlying FD, highlighting the importance of genetic and metabolic factors in disease manifestation.