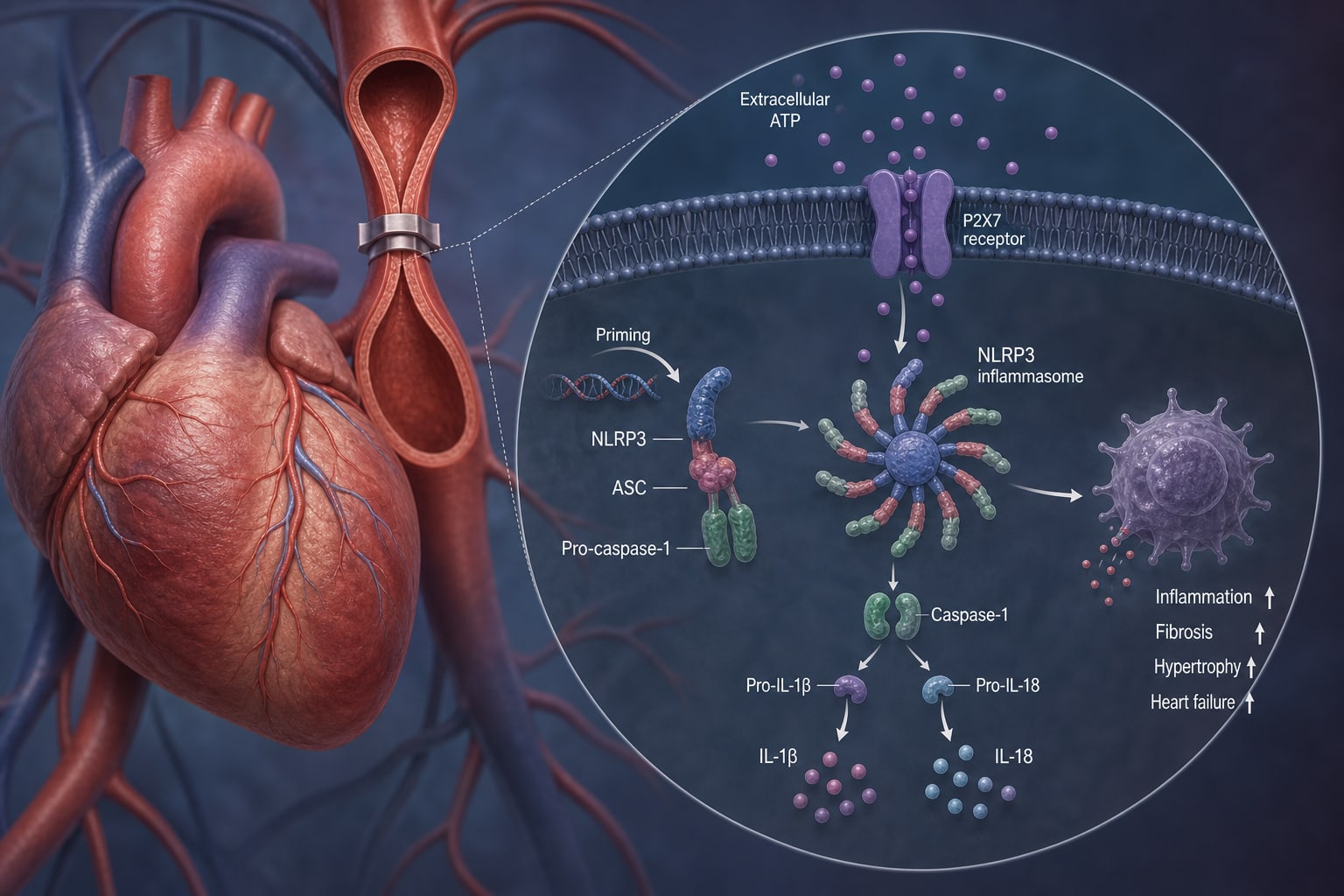
To characterize the stage-specific role of the extracellular ATP–P2X7–NLRP3 inflammasome axis in pressure overload–induced CHF and to assess its therapeutic potential.
Approach:
Model Establishment: A transverse aortic constriction (TAC) mouse model was established and followed longitudinally using hemodynamic assessment and echocardiography.
Evaluation Methods: Extracellular ATP signaling, P2X7 activation, NLRP3 inflammasome priming and assembly, downstream effector responses, histologic remodeling, and inflammatory cell infiltration were evaluated across disease stages.
Causality Testing: Causality was tested using pharmacologic inhibition (P2X7 antagonism and the NLRP3 inhibitor MCC950) and genetic deletion of Nlrp3.
Key Findings:
Pressure overload induced stage-dependent amplification of extracellular ATP–P2X7 signaling.
Progressive activation of NLRP3 inflammasome pathways was observed.
These changes were associated with macrophage accumulation, worsening fibrosis, and declining cardiac function.
Pharmacologic or genetic disruption of the ATP–P2X7–NLRP3 axis attenuated inflammasome signaling, reduced adverse remodeling, and improved cardiac structure and function.
Interpretation:
Extracellular ATP–P2X7 signaling is implicated in NLRP3 inflammasome activation during pressure overload–induced CHF.
Conclusion:
The ATP–P2X7–NLRP3 axis may represent a target for further investigation in hypertension-related CHF.